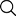Conducting a clinical trial is a complex and challenging task and involves robust scientific understanding and logistics planning.
Although there are international guidelines for good clinical practices, the standard approach may not be suitable for all clinical trials, particularly in cases involving trials that use orphan drugs, patients with terminal illnesses, epidemiological studies, and others.
Factors That Plague Clinical Trials
It has been reported that approximately 50% ofPhase III clinical trials do not achieve their objectives or fail to demonstrate the desired results.
Some of the major issues that pharmaceutical companies face while conducting large-scale trials are:
Meeting regulatory deadlines: Inadequate or poor patient recruitment, poor execution, or complicated study design are some reasons that contribute to the inability of a company to meet timelines. Approximately 80% of trials are behind schedule. Analysis shows patient recruitment to be one of the prime reasons for the study delay.
Data quality: A flawed study design and complacency in following the patient eligibility criteria required for enrolment also affect the data quality and ethics of a trial. In addition, lack of patient informed consent or breach of confidentiality are other serious unethical practices that affect final data quality.
Infrastructure and resources: While accounting for infrastructure and resources, sponsor companies sometimes underestimate the requirement of trained staff at each step of a trial. A sponsor may need to recruit a larger number of clinical trial associates, study coordinators, as well as other trained personnel, depending on the number of trial sites and targeted cohort size.
At times, the importance of site inspection is also overlooked. Site inspections help in evaluating the technical capability of the staff and confirm if the site is well equipped to handle additional responsibilities.
Unexpected challenges: Sponsor companies are sometimes caught unaware by challenges that crop up during the execution of a trial. Without a Risk Management Plan (RMP), it is impossible to identify warning signs, and it can also bring the trial to an abrupt halt.
All the above factors require the sponsor to look for remediation measures, and this is where rescue trials come into the picture.
Rescue Trials
There are different approaches that pharmaceutical companies employ for rescue support.
For specific issues, the company may choose to bring on board a third party with expertise in a specific function or completely outsource study management and control to a contract research organization (CRO).
Integrating into an ongoing study requires the onboarding team to have the flexibility as well as the insight to identify problem areas that have led to the failure of the Sponsor’s trial.
Therefore, it is necessary for the CRO to have demonstrated expertise in handling a particular therapeutic area or to have the technical know-how of running rescue studies.
This will help in seamless knowledge transition and identification of bottlenecks that have caused the trial to fail.
In case the trial is being transferred from another CRO, there should be a clear communication and handover plan from the outgoing CRO to the onboarding CRO and the Sponsor Company.
This communication should include strategic details such as current study status, vendors involved, database migration, documentation, quality control, and current risk management plan, to name some of them.
For a successful rescue study, there should be documented compliance and effective documented communication between the sponsor and CRO.
Corrective action and preventive action (CAPA) at each stage of the trial are necessary, especially in the case of rescue studies, to meet study milestones and to avoid any further delay in trial execution.
Disclaimer:
The information contained in this article is intended solely to provide general guidance on matters of interest for the personal use of the reader, who accepts full responsibility for its use.
Accordingly, the information in this article is provided with the understanding that the author(s) and publisher(s) are not herein engaged in rendering professional advice or services.
As such, it should not be used as a substitute for consultation with a competent adviser. Before making any decision or taking any action, the reader should always consult a professional adviser relating to the relevant article posting.
While every attempt has been made to ensure that the information contained in this article has been obtained from reliable sources, Veeda Lifesciences is not responsible for any errors or omissions or for the results obtained from the use of this information.
All information in this article is provided “as is”, with no guarantee of completeness, accuracy, timeliness, or of the results obtained from the use of this information, and without warranty of any kind, express or implied, including, but not limited to warranties of performance, merchantability, and fitness for a particular purpose.
Nothing herein shall, to any extent, substitute for the independent investigations and the sound technical and business judgment of the reader. In no event will Veeda Lifesciences, or its partners, employees, or agents, be liable to the reader or anyone else for any decision made or action taken in reliance on the information in this article or for any consequential, special, or similar damages, even if advised of the possibility of such damages.
No part of this publication may be reproduced, stored in a retrieval system, or transmitted in any form or by any means, mechanical, electronic, photocopying, recording, or otherwise, without the prior written permission of the publisher.
For information, contact us at:
Veeda Clinical Research Limited
Vedant Complex, Beside YMCA Club, S. G. Highway,
Vejalpur, Ahmedabad – 380 051,
Gujarat India.
Phone: +91-79-3001-3000
Fax: +91-79-3001-3010
Email: info@veedalifesciences.com
Veeda, through its V-Konnect series, interacted with Dr. Arun Bhatt and discussed “Clinical Trials in India and its regulatory perspective with New CT rules.”
About the V- Konnect
V-Konnect interview series is a program to get in touch with specialized industry experts to know their views on opinions on current relevant subject matters.
About Dr. Arun Bhatt – Consultant – Clinical Research & Development
Dr. Bhatt is currently working as a consultant in pharmaceutical medicine and clinical pharmacology.
He is the Editor-in-Chief of the ISCR journal- Perspectives in Clinical Research. Dr. Bhatt has extensive experience in the Indian pharmaceutical industry in clinical research, drug development, and regulatory affairs.
He was the former President of ISCR, President at Clininvent, CEO of CMI (India experience), and Medical Director at Novartis.
He was awarded the ISCR Special Award in 2017 for his contribution to the Research Fraternity, the Outstanding Service award by the Drug Information Association in 2012, and the Honorary Fellowship by Clinical Research UK in 2009.
He is also a qualified assessor of NABH Accreditation for Clinical Trials sites and has more than 150 publications in national and international journals.
Transcript.
1. What are the challenges you see in the Indian clinical trial sector today? Can you please mention how to overcome these challenges?
A:
Quality of trial conduct.
Gaps in Knowledge of Regulations, Ethics, and Science.
Challenges can be tackled if all stakeholders participate passionately in the training and development of their teams and strive for quality in the conduct of clinical trials. Some of the actions are discussed below.
2. Despite professional competence and a large patient pool availability, India is yet to reach its potential in clinical trials. How this can be improved?
A:
Professional competence is not in clinical research but in clinical diagnosis. We need investigators who are trained and passionate about academic clinical research, and who are willing to get trained in clinical trial regulations, ethics, and science, and who are willing to devote time to conduct good clinical research of internationally accepted standards. This is essential to build quality in clinical trials.
Large patient pool is not organized or classified. The sites should develop a detailed database of patients, including demography, disease, and drug information. This would help in quick screening and fast recruitment of subjects.
The government should create awareness about 1) the need for new drug development and clinical trials, and 2) regulatory efforts to ensure the protection of the rights, safety, and well-being of patients.
Ethics committees should receive support, guidance, and training from government bodies – ICMR – and hospital administration to ensure that they can fulfill their primary responsibility of protecting the rights, safety, and well-being of patients.
Industry sponsors should invest in supporting all the above efforts and encouraging academic investigator-initiated trials.
3. With the recent changes in new CT rules, what can be the benefits and shortcomings for clinical trials conducted in India?
A:
Benefits
Time-bound approvals for clinical trials in 90 working days
Advantages of Indian R&D discovery for the initiation of Phase I in 30 working days
Accelerated approval/trial waivers for serious and rare diseases
Challenges:
Investigators: Academic trials to comply with ICMR guidelines , Ethics committees (EC):
Dual registration from the DCGI office and the Department of Health Research Composition: 50% non-affiliated members
Short comings
Independent Non-institutional ECs, which may not be competent in ethical oversight, are permitted to oversee clinical trials
Sponsor is concerned about delays in approval because of irrelevant queries
Lack of clarity/transparency in the regulatory inspection process
No change/improvement in the SEC review process
Approval requirements for non-interventional Phase IV studies
Some of the criteria for accelerated approval/waivers are unclear and are at the discretion of regulatory authorities
4. What are the industry expectations from Indian regulatory going forward?
A:
Transparency and clarity in accelerated/waiver criteria/pathways
Professional regulatory inspection with graded regulatory actions along the lines of the FDA and EMA
5. Where do you see the Indian Clinical trials industry in the next 5 years?
A:
Depends on how the new regulations improve the quality and conduct of Indian trials and how the society and media react to the favorable regulatory environment for new drugs and clinical trials
All stakeholders should learn from past deviations and watchfully conduct clinical trials in compliance with regulations.
Over the next 5 years, all stakeholders should strive for ensuring human protection and data integrity and to establish an image of India as a quality innovation R&D hub.
Focus should be on quality, and Quantity will follow.
6. What are the current issues surrounding clinical trial data integrity, and what can be done to improve it?
A:
Attitudinal shift by sponsors to reject data whose integrity is suspect
Strengthen the QA and monitoring process
Reward sponsor team members who discover data integrity issues and whistleblowers
Action – suspension of the contract, blacklisting, regulatory notification, information sharing with industry – against responsible parties – in-house staff, CROs, investigator sites
Training in documentation, monitoring, and QA for the sponsor and investigator site staff
Training of ECs in oversight and monitoring to detect data integrity issues and to take appropriate actions
7. What measures should the industry take to ensure clinical trials are carried out safely?
A:
Training of in-house staff – monitors, project managers, medical monitors, auditors, and site staff in pharmacovigilance, assessment of causality and clinical trial relationship of SAE, regulatory reporting, and compensation
Train the site personnel thoroughly in protocol procedures, especially selection criteria, follow-ups, and safety assessments
The project team should promptly detect important protocol deviations, which can impact the safety of subjects, and take appropriate actions, e.g., exclude patients, stop recruitment, and inform EC, etc.
Medical monitor and the project team should verify the assessment of causality and clinical trial relationship of SAE by the investigator, considering company safety information and medical condition
Ensure that the investigator complies with the regulatory requirements of free medical management
8. How do you position Indian ethics committees with respect to functioning and competence in the current global scenario? What are your views on the steps to be taken to further improve the functions of the ethics committee?
A:
Barring a few empowered ECs in major academic institutions, most ECs lack competence in fulfilling their vital responsibility of ensuring the protection of rights, safety, and well-being of clinical trial participants.
Department of Health Research should provide training and conduct ongoing monitor/review of the functioning of ECs.
On a closing remark, Dr. Arun Bhatt mentioned that “As a country, we should remember those who forget history are condemned to repeat it, and conduct clinical trials balancing the twin requirements of human protection and data integrity. We should comply with regulations and guidelines both in letter and spirit!”
Disclaimer:
The opinions expressed in this publication are those of the Interviewee and are not intended to malign any ethic group, club, organization, company, individual, or anyone or anything. Examples of analysis performed within this publication are only examples.
They should not be utilized in real-world analytic products as they are based only on the personal views of the Interviewee.
They do not purport to reflect the opinions or views of the VEEDA CRO or its management. Veeda CRO does not guarantee the accuracy or reliability of the information provided herein.
Many respiratory diseases have been historically treated using inhalation drugs, as this route of administration allows for a higher drug concentration to reach the target organ, thereby reducing systemic effects.
Apart from respiratory disorders, trials are ongoing to determine the efficacy of inhaled insulin in diabetes management. 1, 2
Although pulmonary delivery of insulin is a valuable option with the advantage of ease of administration compared to injections, further research is ongoing to study its safety through the oral route. 1, 2
An ideal inhalation device is one that delivers a reproducible and fixed dose of the drug to the lung, is patient-friendly, and is not cumbersome.
The commonly prescribed inhalation devices are pressurized metered-dose inhalers (MDIs), nebulizers, and dry-powder inhalers.
All inhalation devices undergo stringent in vivo and in vitro testing to determine the safety and efficacy of the drug through these devices. 3
However, inhalation clinical trials bring forth a number of challenges.
Device-drug Compatibility
There are hiccups that investigators and sponsors face while conducting inhalation trials, such as the need to use cumbersome and costly devices as well as the probability of bronchospasm due to the drug or non-drug component(s).
In addition, some inhalation drugs can cause withdrawal symptoms.
Other factors that influence the trial results are the difference in drug bioavailability in each patient due to varied breathing patterns or the presence of a comorbidity that affects drug absorption.
For instance, epoprostenol has a short half-life of 3 to 5 minutes, requiring continuous nebulization for long periods, making it difficult to administer or prescribe on a long-term basis.3
Safety Issues
A number of inhalation trials were terminated in the initial phases due to issues such as poor drug solubility and bioavailability, leading to dangerous levels of undissolved drug in the systemic circulation. 4
Patient Training and Adaptability
For effective therapy, the patient should be able to use the device correctly.
Inhalation drugs and devices are often viewed as complex by many patients, and thus require practical demonstration as well as repeated follow-up by medical staff to ensure that the patient is using the device as intended for optimal drug delivery.
The patient should also be encouraged to use e-technologies that help in self-monitoring to look out for symptoms that may require medical intervention and help raise awareness about the respiratory disease. 5
Many studies have observed that the inappropriate use of inhalers is a cause for improper management of respiratory diseases.
A study in a university hospital in Northwest Ethiopia by Mebrahtom Metal demonstrated that approximately 71% of the subjects were handling inhalation devices incorrectly due to a lack of awareness about MDIs, consequently leading to poor asthma control. 6
Another study by Arora Petal reported approximately 95% error in subjects using MDI and approximately 82% error in subjects using dry powder inhalers. 7
Regulatory Laws in India
There are no specific regulatory guidelines laid down by the legislative body, Central Drugs Standard Control Organization (CDSCO), and the Drug Controller General of India (DCGI), for inhaled products.
As applicable to all trials in India, inhalation clinical trials should also adhere to Schedule Y and Rules 122A to E of the Drugs and Cosmetics Act, 1945, as well as Good Clinical Practices (GCP) and ethical guidelines for biomedical research on human subjects.
The guidelines followed for bioavailability and bioequivalence studies are also applied to inhalation trials.
However, bioequivalence studies for inhaled drugs are still in their nascent stages in India.
Although pharmacokinetic (PK) bioequivalence studies alone are being accepted worldwide to establish equivalence of inhalation products, India is yet to approve second entry orally inhaled drugs with data from PK bioequivalence studies alone. 8
Creating a Conducive Environment for Inhalation Clinical Trials
To build India’s competence in inhalation trials, the recruited staff should have expertise in handling phase I/bioequivalence studies.
The DCGI and CDSCO can also come up with specific study timelines as well as suggest appropriate study designs for inhalation trials in consultation with the technical committee.
As the comparison of clinical efficacy endpoints between two orally inhaled products provides shallow dose response curves, equal weightage should be given to in vitro bioequivalence assessments. 8
Emphasis cannot be laid enough on the need for the investigator to share all medical decisions with the patient to improve patient compliance rates in clinical trials. 5
Human factor (HF) studies can be designed to include strategies that mitigate errors caused by improper device use.
HF studies also help in understanding the effect of the interaction between the patient and device on the safety and efficacy of the inhalation drug.
HF studies are gradually showing their presence globally, especially in clinical trials that involve the use of devices. 9
With companies increasingly seeking alternative solutions, the route of inhalation delivery will continue to grow.
This makes it critical for the scientific community to fill the existing gaps for conducting successful inhalation trials.
Sources
Cavaiola TS and Edelman S. Inhaled Insulin: A Breath of Fresh Air? A Review of Inhaled Insulin. Clinical Therapeutics. 2014;36(8):1275-89.
Oleck J, Kassam S, and Goldman JD. Commentary: Why Was Inhaled Insulin a Failure in the Market. Diabetes Spectrum. American Diabetes Association. 2016;29(3):180-4. https://doi.org/10.2337/diaspect.29.3.180
Holgate ST, Bousquet J, Chung KF et al. Summary of recommendations for the design of clinical trials and the registration of drugs used in the treatment of asthma. Respiratory Medicine, 2004;98(6):479–487.
Forbes B, O’Lone R, Allen PP et al. Challenges for inhaled drug discovery and development: Induced alveolar macrophage responses. Advanced Drug Delivery Reviews. 2014;71:15-33.
Shakshuki A and Agu RU. Improving the Efficiency of Respiratory Drug Delivery: A Review of Current Treatment Trends and Future Strategies for Asthma and Chronic Obstructive Pulmonary Disease. Pulmonary therapy. 2017;3:267-81.
Mebrahtom M, Mesfin N, Gebreyesus H et al. Status of metered dose inhaler technique among patients with asthma and its effect on asthma control in Northwest Ethiopia. BMC research notes. 2019;12:15.
Arora P, Kumar L, Vohra V et al. Evaluating the technique of using inhalation device in COPD and bronchial asthma patients. Respiratory Medicine. 2014;108(7):992-8.
Lee SL, Saluja B, Garcia-Arieta A et al. Regulatory Considerations for Approval of Generic Inhalation Drug Products in the US, EU, Brazil, China, and India. AAPS Journal. 2015;17(5):1285-1304.
The information contained in this article is intended solely to provide general guidance on matters of interest for the personal use of the reader, who accepts full responsibility for its use.
Accordingly, the information in this article is provided with the understanding that the author(s) and publisher(s) are not herein engaged in rendering professional advice or services.
As such, it should not be used as a substitute for consultation with a competent adviser. Before making any decision or taking any action, the reader should always consult a professional adviser relating to the relevant article posting.
While every attempt has been made to ensure that the information contained in this article has been obtained from reliable sources, Veeda Lifesciences is not responsible for any errors or omissions or for the results obtained from the use of this information.
All information on this article is provided “as is”, with no guarantee of completeness, accuracy, timeliness or of the results obtained from the use of this information, and without warranty of any kind, express or implied, including, but not limited to warranties of performance, merchantability and fitness for a particular purpose.
Nothing herein shall to any extent substitute for the independent investigations and the sound technical and business judgment of the reader.
In no event will Veeda Lifesciences, or its partners, employees, or agents, be liable to the reader or anyone else for any decision made or action taken in reliance on the information in this article or for any consequential, special, or similar damages, even if advised of the possibility of such damages.
No part of this publication may be reproduced, stored in a retrieval system, or transmitted in any form or by any means, mechanical, electronic, photocopying, recording, or otherwise, without the prior written permission of the publisher.
In the clinical research industry, safety norms and processes should be applied to every aspect, starting from drug development to post-marketing use of approved drugs, keeping in mind patient safety and data credibility at every stage of clinical development.
Patient safety requires the collaborative effort of the regulatory system, the healthcare system, and the sponsor.
Equally important is the need for effective communication with patients by the regulatory bodies and health care systems to maintain transparency.
Such communication also helps in instilling confidence in patients about the scientific credibility of the trial.
All data, starting from the filing of an application to the trial results, is available on the website.
Sponsors, clinical research organizations, as well as ethics committees are required to furnish their details on the website.
Data safety and monitoring boards (DSMB) are also formed to monitor trials, especially large trials, for safety and reliability of data.
DSMBs play a significant role in letting the public know if the investigational drug poses any threat, as well as to ensure that the sponsor does not purport the results to be unbelievably beneficial to the targeted patient population.
Emphasis should also be laid on novel and practical-oriented training programs for students in the health care profession to expose them to real-world situations.
Students should understand the importance of having risk assessment, risk management, and risk mitigation plans in place to improve patient safety.
Periodic assessment and continuing medical education programs for medical professionals are also pivotal in ensuring that they remain competent in their respective fields and prioritize patient safety and moral ethics in their profession.
The successful implementation of patient safety and data integrity is a multidisciplinary approach that requires sincere and diligent effort of each individual and organization involved in the conduct of clinical trials.
Disclaimer:
The information contained in this article is intended solely to provide general guidance on matters of interest for the personal use of the reader, who accepts full responsibility for its use.
Accordingly, the information in this article is provided with the understanding that the author(s) and publisher(s) are not herein engaged in rendering professional advice or services.
As such, it should not be used as a substitute for consultation with a competent adviser. Before making any decision or taking any action, the reader should always consult a professional adviser relating to the relevant article posting.
While every attempt has been made to ensure that the information contained on this article has been obtained from reliable sources, Veeda Lifesciences is not responsible for any errors or omissions, or for the results obtained from the use of this information.
All information on this article is provided “as is”, with no guarantee of completeness, accuracy, timeliness or of the results obtained from the use of this information, and without warranty of any kind, express or implied, including, but not limited to warranties of performance, merchantability and fitness for a particular purpose.
Nothing herein shall to any extent substitute for the independent investigations and the sound technical and business judgment of the reader.
In no event will Veeda Lifesciences, or its partners, employees or agents, be liable to the reader or anyone else for any decision made or action taken in reliance on the information on this article or for any consequential, special or similar damages, even if advised of the possibility of such damages.
No part of this publication may be reproduced, stored in a retrieval system, or transmitted in any form or by any means, mechanical, electronic, photocopying, recording, or otherwise, without the prior written permission of the publisher.
For information, contact us at:
Veeda Clinical Research Limited
Vedant Complex, Beside YMCA Club, S. G. Highway,
Vejalpur, Ahmedabad – 380 051,
Gujarat India.
Phone: +91-79-3001-3000
Fax: +91-79-3001-3010
Email: info@veedalifesciences.com
The country offers the advantages of having experienced personnel, good infrastructure such as well-equipped hospitals and laboratories, and a diverse patient pool.
Moreover, the increase in life expectancy rate to 65 years and above gives rise to a host of lifestyle diseases such as diabetes, hypertension, neurodegenerative diseases, and more, paving the way for many multinational pharmaceutical companies to invest in clinical trials in India.
The recent amendment made by the Ministry of Health and Family Welfare, Government of India, to the regulatory laws (New Drugs and Clinical Trials Rules, 2019) was with the aim of increasing the percentage of clinical trials conducted in India, which hit an all-time low in the period between 2011 and 2013.
In a bid to streamline approval processes, well-defined timelines have been introduced.
The new rules have shortened the approval timeline for clinical trials of drugs manufactured outside India to 90 days.
For drugs manufactured in India, the clinical approval timeline is 30 days.
Pre-submission and post-submission meetings between the sponsor and the Drugs Controller General of India (DCGI) are attempts to increase transparency in all dealings related to the clinical trial.
In addition, the non-refundability clause of the compensation package was removed as this was seen as a major deterrent for international companies, especially if a death/injury was proved to be unrelated to the trial at later stages.
Apart from clinical trials, there has been a boom in the field of data management and medical writing, with a number of homegrown clinical research organizations (CROs) demonstrating expertise to handle end-to-end services for sponsor companies.
Last but not least, concerted efforts and joint participation of the Indian government and Indian pharmaceutical companies in policy-making decisions and prioritization of patients’ safety and health will build confidence in international companies about India’s capability in contributing to the clinical research industry.
Shaping of a Regulatory Framework Specific to the Indian Clinical Market
As India emerges as one of the leaders in the production of generic pharmaceuticals, contributing to approximately 20% of the global market 1 , necessary to have regulatory authorities approve a larger number of drugs or clinical trials to address the burden of diseases prevalent in India.
However, to safeguard public health, it is equally important to ensure that national and international pharmaceutical companies comply with stringent regulatory processes laid down for the approval of drugs.
Changes in the Indian Regulatory Scenario
Revision of Schedule Y of the Drugs and Cosmetics Act 1945 in 2005 helped in aligning the Indian regulatory framework with internationally accepted definitions and procedures.
In addition, the Indian Good Clinical Practices (GCP) guidelines that were drafted by the expert committee of the Central Drugs Standard Control Organization (CDSCO) helped in ensuring uniformity in the conduct and quality of clinical research across the country.
With the recent introduction of the New Drugs and Clinical Trial Rules by the Ministry of Health and Family Welfare of India in 2019, the government is focusing on fast-tracking the approval of new drugs, with equal weightage given to bio-equivalence or bioavailability studies. 2
The government has also ensured further strengthening of the regulatory sector by allocating a higher budget of approximately 65 million US dollars at the central and state levels.
This has helped in improving the infrastructure, such as setting up an E-governance portal (SUGHAM) to bring in transparency, accountability, and ease of business.
A notable initiative by the CDSCO was the establishment of a pharmacovigilance program in 2010, aimed at implementing robust systems for adverse event reporting. 1
The New Regulatory Rules
Some of the regulatory rules that have become effective following the New Drugs and Clinical Trials Rules 2019 are:
Timelines for the approval of global clinical trial applications have been revised to 90 days, and for domestic trials, to 30 days, from the previous duration of 6 months. 3
Phase III clinical trials are not required for any new drugs that have been approved for sale in the United States, Canada, Australia, or the United Kingdom. 3
Post-marketing surveillance studies are to be conducted in India to monitor for idiosyncrasies or unexpected adverse events.3
Participant has access to a free drug post-trial if no suitable alternative is available in the market. However, the Sponsor would not be responsible for any complications that occur post the study duration.4
The approval obtained for a clinical trial is valid for 2 years.4
Compensation for death/injury/disability that is related to the trial will be decided by the Drug Controller General of India (DCGI).4
Conclusion
To head towards India’s goal of becoming a competent clinical trial destination, it is important to not only speed up the approval of drugs or clinical trials but also to keep allegiance to the Ethics committee and to the Indian Council of Medical Research’s (ICMR) National Ethical Guidelines for Biomedical and Health Research involving Human Participants, to prevent exploitation of trial participants.5
Transparency in the drug approval process and stringent laws with penalties for unethical conduct of trials can become game changers with respect to maintaining high standards of quality for drugs and reliability of data from clinical trials.
The regulatory environment can also see an improvement in the implementation of regulatory laws by increasing the number of skilled staff, such as drug inspectors, regulatory specialists, and so on.
Equal emphasis should be laid on standard operating procedures (SOPs) and updating guidance documents to help staff understand the current regulatory environment.
Exchange programs with other countries that have sound regulatory processes can be beneficial for the Indian regulatory personnel to appreciate the importance of a well-defined regulatory framework and how some aspects can be practically implemented in the Indian regulatory environment. 6
Of utmost priority should be a patient’s safety and rights. Having a representative from patient advocacy groups in important decision-making meetings held by the DCGI allows for a patient-centric approach with respect to the review of policies and laws.6
To conclude, high-quality drugs and ethical clinical trials are the joint responsibility of sponsors, clinical investigators, and regulatory bodies.
Periodically updating the Indian regulations can address loopholes and instill confidence in international pharmaceutical companies to continue investing in India. This will, in turn, propel India’s economic growth.
Sources
Medicine regulation. Regulatory systems in India. WHO Drug Information. 2017;31(3). https://www.who.int/medicines/publications/druginformation/issues/WHO_DI_31-3_RegSystemIndia.pdf?ua=1
The information contained in this article is intended solely to provide general guidance on matters of interest for the personal use of the reader, who accepts full responsibility for its use. Accordingly, the information in this article is provided with the understanding that the author(s) and publisher(s) are not herein engaged in rendering professional advice or services.
As such, it should not be used as a substitute for consultation with a competent adviser. Before making any decision or taking any action, the reader should always consult a professional adviser relating to the relevant article posting.
While every attempt has been made to ensure that the information contained in this article has been obtained from reliable sources, Veeda Lifesciences is not responsible for any errors or omissions or for the results obtained from the use of this information.
All information in this article is provided “as is,” with no guarantee of completeness, accuracy, timeliness, or of the results obtained from the use of this information, and without warranty of any kind, express or implied, including, but not limited to warranties of performance, merchantability, and fitness for a particular purpose.
Nothing herein shall, to any extent, substitute for the independent investigations and the sound technical and business judgment of the reader.
In no event will Veeda Lifesciences, or its partners, employees, or agents, be liable to the reader or anyone else for any decision made or action taken in reliance in the information on this article or for any consequential, special, or similar damages, even if advised of the possibility of such damages.
No part of this publication may be reproduced, stored in a retrieval system, or transmitted in any form or by any means, mechanical, electronic, photocopying, recording, or otherwise, without the prior written permission of the publisher.
For information, contact us at:
Veeda Clinical Research Limited
Vedant Complex, Beside YMCA Club, S. G. Highway,
Vejalpur, Ahmedabad – 380 051,
Gujarat India.
Phone: +91-79-3001-3000
Fax: +91-79-3001-3010
Email: info@veedalifesciences.com
Advancement in medical sciences have benefited humanity in many ways.
However, in the process of conducting clinical trials, incidences of scientific, moral, and ethical misconduct have been unearthed that have shaken up the scientific community and the public.
This led to the formation of a formal organization in 1979 by the United States (US), namely the “International Ethical Guidelines for Biomedical Research Involving Human Subjects”, to protect and safeguard the interests of trial subjects.
Following this, many countries drafted their own guidelines for Good Clinical Practices (GCP).
However, with the increasing number of clinical trials being conducted at sites in multiple countries, it was necessary to have a uniform guideline for conducting clinical trials.
This gave rise to the International Conference on Harmonization (ICH)-GCP guidelines in 1996 with the objective of providing a uniform standard that facilitates the acceptance of clinical trial data by the regulatory authorities of the respective countries.
Over the course of time, many countries adapted the ICH-GCP guidelines to frame their own guidelines.
India too followed suit with the Indian Council of Medical Research (ICMR) introducing the “Ethical Guidelines for Biomedical Research on Human Subjects,” which is continuously revised and amended to ensure that clinical trials are conducted with utmost quality, giving priority to the welfare of the subjects involved.
India – A Global Destination
India is emerging as a favorite destination for clinical trials for many international companies due to several factors:
Conducive Regulatory Environment: Internationally harmonized and favorable regulatory processes, such as fast-track approval of investigational new drugs, make the Indian clinical research environment more amenable to conducting clinical trials. Market trends show a compound annual growth rate (CAGR) of approximately 12% (US dollars 987 million) in the Indian clinical trials industry from US dollars 500 million in 2017.
Trained Manpower: Availability of skilled healthcare professionals who are specialists in different therapy areas, well-versed in the English language, and who ensure compliance with ICH-GCP guidelines.
Technology Infrastructure: World-class technologies in data management, information technology, and related services.
Patient Pool: Large population who are treatment naïve and have a diverse genetic and ethnic makeup. With India becoming increasingly urbanized and with greater connectivity between the urban and rural areas, it becomes convenient to recruit patients from different geographical areas. In addition, there is a high incidence and prevalence of acute and chronic diseases due to lifestyle changes, leading to diseases such as diabetes, cancer, and so on. Such lifestyle-related disorders open up the possibility of conducting more clinical trials in these disease areas.
Ease of recruitment: In countries such as the US, approximately 86% of the clinical trials fail to recruit the required number of subjects, leading to a delay of almost a year. This delay costs the sponsor company several million dollars. Some of the reasons for delayed recruitment are unwillingness of patients to participate, stringent safety requirements, and hefty compensation packages. India provides the possibility of the recruitment of patients with relative ease due to increased trial compliance and transparency, especially with the recent release of the New Drugs and Clinical Trial Rules 2019, which consists of updated rules and regulations for fast-tracking approval of clinical trials. Among countries with fast-growing economies, it has been noted that India has a growth rate in recruitment sites of approximately 22.6%, with the highest growth rate seen in China (≈36%).
Competitive costs – Cost effectiveness is a driving factor for many trials being shifted to India. The cost to develop a new drug is estimated to be almost 50% less than what would be required in the US or in the European Union.
Future of Clinical Research in India
Specific guidelines are being worked upon by the Central Drugs Standard Control Organization (CDSCO) for stem cell research, biosimilars, and medical devices to protect patients as well as to encourage clinical research and development in the country.
After a lull period in the Indian clinical industry before 2015 due to ethical and quality concerns, open communication between sponsor representatives and the regulatory team of CDSCO has helped in reconsidering India once again as a potential global destination for enrolling a diverse population in clinical trials that adhere strictly to ICH-GCP guidelines.
Disclaimer:
The information contained in this article is intended solely to provide general guidance on matters of interest for the personal use of the reader, who accepts full responsibility for its use.
Accordingly, the information in this article is provided with the understanding that the author(s) and publisher(s) are not herein engaged in rendering professional advice or services.
As such, it should not be used as a substitute for consultation with a competent adviser. Before making any decision or taking any action, the reader should always consult a professional adviser relating to the relevant article posting.
While every attempt has been made to ensure that the information contained in this article has been obtained from reliable sources, Veeda Lifesciences is not responsible for any errors or omissions or for the results obtained from the use of this information.
All information on this article is provided “as is”, with no guarantee of completeness, accuracy, timeliness or of the results obtained from the use of this information, and without warranty of any kind, express or implied, including, but not limited to warranties of performance, merchantability and fitness for a particular purpose.
Nothing herein shall to any extent substitute for the independent investigations and the sound technical and business judgment of the reader.
In no event will Veeda Lifesciences, or its partners, employees, or agents, be liable to the reader or anyone else for any decision made or action taken in reliance on the information in this article or for any consequential, special, or similar damages, even if advised of the possibility of such damages.
No part of this publication may be reproduced, stored in a retrieval system, or transmitted in any form or by any means, mechanical, electronic, photocopying, recording, or otherwise, without the prior written permission of the publisher.
For information, contact us at:
Veeda Clinical Research Limited
Vedant Complex, Beside YMCA Club, S. G. Highway, Vejalpur, Ahmedabad – 380 051, Gujarat, India.
Phone: +91-79-3001-3000
Fax: +91-79-3001-3010
Email: info@veedalifesciences.com
Biosimilars or biogenerics are biopharmaceutical products with large and complex structures that are similar to their innovator products in all aspects, including safety, efficacy, and quality.
There is a palpable sense of urgency among researchers and the pharmaceutical industry to develop biosimilars for treatment, with their popularity expected to rise in the forthcoming years, especially in developing low-cost biosimilars that are easily accessible and affordable for patients.
Market trends indicate that North America remains the dominant force in the global oncology biosimilar market, followed by Europe, the Asia Pacific, Latin America, the Middle East, and Africa.
The market is expected to reach approximately USD 45 billion by 2026, up from the current market of approximately USD 6 billion.
Understanding the local and global market demand is necessary for companies, research institutes, and investors to study the changing dynamics in the biosimilar market.
Segmentation of biosimilars by product, type of cancer, as well as targeted end-users such as hospitals/online/retail pharmacies is particularly useful in empowering industries to make informed business decisions.
Product segmentation also helps gauge the project’s feasibility so that the pharmaceutical industry can focus on developing safe and commercially viable products.
Comprehensive analyses, robust legal and regulatory frameworks, continuing medical education for healthcare professionals, and increased awareness among the public will undoubtedly increase the uptake of biosimilars and help the biosimilar market move forward competitively.
The scientific community has been a witness to some of the worst tragedies in the history of clinical trials data integrity.
From the year 2015 to date, The Journal of the American Medical Association (JAMA) and the JAMA Network journals have published at least 18 notices citing concern over data error and/or falsification of data.
For instance, the trials conducted by a Japanese anesthesiologist and researcher to treat post-operative nausea and vomiting were reviewed by the Japanese Society of Anesthesiologists (JSA) in the year 2012 to find startling revelations. 1
The data obtained from the trials were either totally fabricated or fraudulent, and approximately 210 papers published by the anesthesiologist had falsified data.
Lapses in data integrity resulted in a significant loss of revenue, with direct costs estimated to be nearly $525,000 US dollars, while indirect costs totaled approximately $1.3 million US dollars. 2, 3
Such scientific misconduct served as a wake-up call to tighten regulations and laws to monitor drug development and drug use.
Scientists acknowledged the need for data integrity at every stage to safeguard human subjects, starting from pre-clinical development to pharmacovigilance.
What is Data Integrity?
Data integrity is defined as paper-based or electronic data that is complete, accurate, consistent, and reliable through its lifecycle from the time of data creation, archival, scanning, retention, and destruction.
The updated International Council for Harmonization Guideline for Good Clinical Practice (ICH GCP E6[R2]) reiterates the need for data integrity as well as the importance of monitoring clinical data throughout the study. 4
The United States Food and Drug Administration (FDA) uses the ALCOA acronym to define expectations with respect to data integrity. 4
Data Compliance Issues
The FDA issued Good Manufacturing Practices (GMP) warning letters to various countries outside the United States, citing compliance issues related to data integrity.
Figure 1 shows that China has received the maximum number of GMP warning letters, followed by India and Europe. 5
Figure 1: GMP warning letters issued outside the US. 5
Data Integrity Checkposts
Data integrity can be monitored by keeping a check on the following areas: 6
►Source Data Verification (SDV)
►Data access and control
►Training of personnel involved in data collection
such as investigators, data processors, analysts, site staff, and report writers
►Data monitoring: On-site, centralized, and risk-based monitoring
►Clinical trial quality assurance units (QAU): Some sponsors set up internal QAUs or external QAUs with a contract research organization (CRO) to ascertain trial compliance with standard operating procedures (SOPs) and FDA regulations. QAUs also eliminate the risk of internal bias. Regulatory laws, however, do not mandate the need for a QAU.7
►Clinical trial audits
SDV
Strict adherence to good documentation practices (GDP) in clinical trial records is a way to ensure data integrity. GDP should be followed for paper records as well as electronic records and signatures.
Equally important is the need to retain and organize essential documents required before the start of a clinical trial, during the trial, and after the completion or termination of a trial.
The collection of essential documents that is kept at the sponsor site and investigator site is called the clinical trial master file (TMF).
TMF plays a major role in facilitating trial conduct and management, thereby allowing for data integrity and GCP compliance at all stages of the clinical trial.
The TMF is the document that is reviewed during an audit or inspection.8
It is necessary to exercise caution while handling data from clinical trials. Confidentiality of data should be maintained during all the phases of a clinical trial, including interim data results.
The ability to tamper with data, such as changing, deleting, or falsifying data, should be restricted by clearly demarcating roles. 9
This also prevents potential conflicts of interest between similar roles that may hamper data integrity. 4
The National Institute of Health (NIH) states that only voting members of the Data and Safety Monitoring Board (DSMB) should be permitted to look at the interim analysis results unless circumstances make it necessary to share data, such as in the case of serious adverse events.
In addition, the DMC members should not have any conflict of interest that would influence the outcome data. 9
The FDA has also recommended the use of an “independent statistician” model to analyze interim data, who is independent of the principal investigator and trial sponsor and reports unbiased results to the DMC.10
Data Monitoring
It is necessary to set up an independent data monitoring committee (DMC) that prioritizes the safety and interests of enrolled subjects and scrutinizes the authenticity of data as well as the clinical trial conduct. 9
On-site Monitoring: is carried out to trace any discrepancy between the source data and the data entered. It is also particularly useful to see if the site staff is familiar with the study document and if the staff has demonstrated accountability to carry out the trial ethically and responsibly.11
Centralized Risk-Based Approach: ICH GCP E6(R2) emphasizes the need for centralized monitoring to reduce the number of trial visits by the clinical monitor and to allow for remote spotting of reliable and unreliable data by statisticians or other data management staff. 4,11
Risk-based Monitoring: The sponsor company is required to develop a robust risk management plan to prevent or mitigate any risk to human subjects by overseeing trial conduct and monitoring data quality across trial sites. 11
Data Integrity Audits 12
Specific audits look out for any data or metadata that previously went unnoticed, such as deleted or unchecked, misused, orphaned, or reprocessed data.
The entire data lifecycle should be subjected to scrutiny by all departments involved in the trial, such as but not limited to data management, safety, quality risk management, and statisticians, for compliance issues in areas of data management and data access control.4,8
Unnecessary incentivization for speedy results or data from high-risk phase II trials should be closely monitored for unscrupulous activities.
Weightage should be given to raw data, and not summary reports and results, which should be reviewed for any compliance issues.
Conclusion
To avoid huge financial repercussions and loss of business, sponsor companies and CROs should place sufficient emphasis on maintaining data integrity at every step of the clinical study for its completeness, accuracy, and consistency.
George SL and Buyse M. Data fraud in clinical trials. Clin Investig (Lond). 2015; 5(2): 161–173.
Michalek AM, Hutson AD, Wicher CP et al. The Costs and Underappreciated Consequences of Research Misconduct: A Case Study. PLoS Med. 2018;7(8):e1000318. https://doi.org/10.1371/journal.pmed.1000318
Rutherford M. ICH E6(R2) and Data Integrity: Four Key Principles. Clinical Researcher. 2018 April;32(4):doi:10.14524/CR-18-4021. https://acrpnet.org/2018/04/17/ich-e6r2-data-integrity-four-key-principles/
http://firstclinical.com/fda-gcp/?show=MonitoringvAuditing&search=compliance&type=&page=1 Accessed on April 26, 2019
https://www.ema.europa.eu/en/documents/scientific-guideline/draft-guideline-good-clinical-practice-compliance-relation-trial-master-file-paper/electronic-content-management-archiving-audit-inspection-clinical-trials_en.pdf Accessed on April 26, 2019
Fleming TR, Sharples K, McCall J et al. Maintaining the confidentiality of interim data to enhance trial integrity and credibility. Clin Trials. 2008;5(2):157-67. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2703711/
Ellenberg SS. Protecting Clinical Trial Participants and Protecting Data Integrity: Are We Meeting the Challenges? PLoS Med. 2012 Jun;9(6):e1001234.
The information contained in this article is intended solely to provide general guidance on matters of interest for the personal use of the reader, who accepts full responsibility for its use.
Accordingly, the information in this article is provided with the understanding that the author(s) and publisher(s) are not herein engaged in rendering professional advice or services. As such, it should not be used as a substitute for consultation with a competent adviser.
Before making any decision or taking any action, the reader should always consult a professional adviser relating to the relevant article posting.
While every attempt has been made to ensure that the information contained in this article has been obtained from reliable sources, Veeda Lifesciences is not responsible for any errors or omissions or for the results obtained from the use of this information.
All information in this article is provided “as is,” with no guarantee of completeness, accuracy, timeliness, or of the results obtained from the use of this information, and without warranty of any kind, express or implied, including, but not limited to warranties of performance, merchantability, and fitness for a particular purpose.
Nothing herein shall, to any extent, substitute for the independent investigations and the sound technical and business judgment of the reader.
In no event will Veeda Lifesciences, or its partners, employees, or agents, be liable to the reader or anyone else for any decision made or action taken in reliance on the information in this article or for any consequential, special, or similar damages, even if advised of the possibility of such damages.
No part of this publication may be reproduced, stored in a retrieval system, or transmitted in any form or by any means, mechanical, electronic, photocopying, recording, or otherwise, without the prior written permission of the publisher.
For information, contact us at:
Veeda Clinical Research Limited
Vedant Complex, Beside YMCA Club, S. G. Highway,
Vejalpur, Ahmedabad – 380 051,
Gujarat India.
Phone: +91-79-3001-3000 Fax: +91-79-3001-3010 Email:info@veedalifesciences.com
India is emerging as a country with tremendous potential to contribute to the national and international clinical trial platforms.
The Central Drugs Standard Control Organization (CDSCO) is the national regulatory authority of India, overseen by the Drug Controller General of India (DCGI). 1,2,3
The DCGI is responsible for coordinating inspections of sponsors, manufacturing units, and trial sites. 3
Early Years in Clinical Development
In 2000, the Indian Council of Medical Research (ICMR) set up ethical guidelines for conducting biomedical research on human subjects. 4
The year 2005 saw a revision of Schedule Y of the Drugs and Cosmetics Act, 1945, to align Indian regulatory laws with internationally accepted definitions and procedures.
Demarcated responsibilities of sponsor(s) and investigator(s)
Options to record any deviation or changes to the approved study protocol
India also signed the Trade-Related Aspects of Intellectual Property Rights (TRIPS) agreement in 2005, aiming to open up the prospects for conducting more clinical trials in the country. 5
Apart from harmonizing the regulatory acts to international standards, India quickly became a favorable destination for clinical trials as it offered: 6
English-speaking professionals in health care
Technical expertise
Growing economy
World-class technology
Large, diverse, and treatment-naïve population
Set Back for Clinical Trials
Despite changes in the regulations, many multinational pharmaceutical companies took advantage of the large population that had either inadequate knowledge about clinical trials or was illiterate.
Additionally, an ill-defined healthcare system contributed to the challenges of monitoring unethical practices.
This led to conducting clinical trials with little supervision and no recording of patient-informed consent, either in written or audio/visual form.
Patients were administered investigational drugs or devices without disclosing known serious adverse effects, some leading to the death of the subjects.
Moreover, no independent enquiry committee was set up to ascertain if the death of the patient was related/not to the investigational product or device. 4
The years 2010 to 2013 witnessed a challenging phase in the Indian clinical trial scenario due to the cumulative adverse effects of conducting unethical trials.
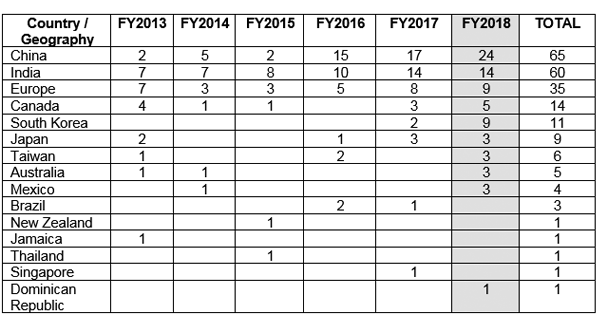
However, with a better regulatory framework in place, the Clinical Trial Registry of India (CTRI) has recorded a steady rise in the number of trials being conducted, as seen in Figure 1.
It was also observed that most of the trials were phase III trials. 7
Figure 1: Clinical trial trends over the years
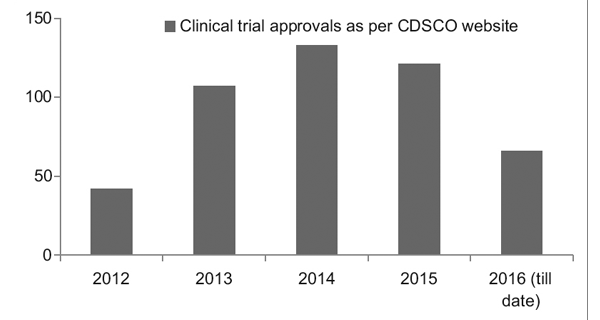
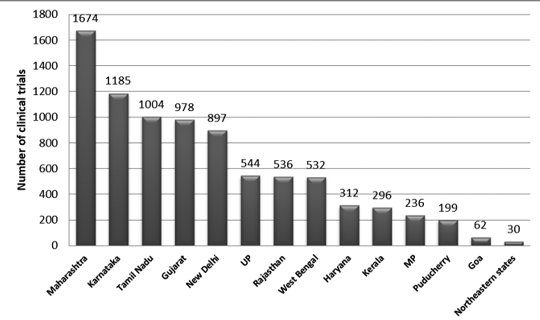
Figure 2 presents the state-wise distribution of trials in India between 2007 and 2015. Approximately 3330 trails were registered during this period.
It was observed that the maximum number of trials was conducted in Maharashtra, and the least number of trials was conducted in the Northeastern state.
Among the Northeastern states, no trials were conducted in Nagaland.7
Figure 2: State-wise distribution of clinical trials in India (2007-2015 data)7
Revival of the Clinical and Regulatory Scenario
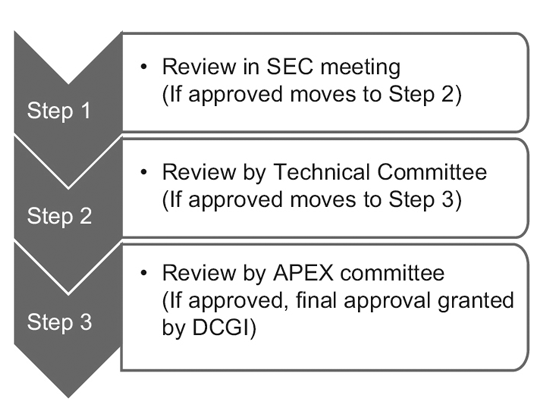
In 2014, the CDSCO constituted 12 new drug advisory committees (NDACs) and 25 subject expert committees (SECs).
These committees comprise experts from eminent government colleges and institutions, aiming to expedite the approval timelines of clinical trials to 6-7 months.
The three-tier process consists of: 9
However, only the SEC reviews global clinical trial applications, and no further approval is required from the Technical Committee or the Apex Committee.
Investigational New Drug (IND) applications are also reviewed independently by the IND committee and do not require the approval of the Apex committee.
A Technical committee comes into the picture only if the SEC has rejected a sponsor’s application and the sponsor feels aggrieved by the decision.
In such an event, if the Technical committee disagrees with the decision of the SEC, it has the power to overrule the decision of the SEC.10
In March 2019, the Ministry of Health and Family Welfare, India, released the New Drugs and Clinical Trial Rules 2019 with the intention of fast-tracking the approval for clinical trials, new drugs, bioequivalence (BE), and bioavailability (BA) studies.
These rules have also addressed any ambiguity that existed with respect to the regulation of the Ethics Committee (EC). 11
Highlights of the New Drugs and Clinical Trial Rules, 201911
Updated rules and regulations 11
Approval timeline for clinical trials
90 working days from receipt of an application for drugs discovered outside India and 30 working days for new drugs or an IND in India
Manufacturing of new drugs or IND, BE & BA studies, or test analysis or examination
Permission is required from the Central Licensing Authority (CLA)
Waiver of local clinical trials
· If CLA has approved the marketing of the new drug in other countries or has granted permission to conduct global clinical trials for the new drug in India
· No evidence of a difference in metabolism, safety, or efficacy owing to the difference in the genetic profile of the Indian population
Period of validity of a clinical trial
2 years from the date of issue by CLA
Post-trial access to an IND or a new drug
In unique circumstances, the drug is to be distributed free of cost to trial subjects per the direction of CLA, but no liability lies with the sponsor for the use of the drug after trial.
Pre-submission and post-submission meetings
To seek guidance with respect to the law and procedures that govern the process of manufacturing, licensing, or granting permission.
Approval for trials conducted by the EC and registration of the EC
· Approval to be obtained from the EC of another trial site if a trial site does not have an EC, and the EC should be within 50 km of the trial site.· CLA-approved registration of EC remains valid for five years from the date of issue.
Conditions to be fulfilled for the conduct of a clinical trial
· Submission of a status report on a quarterly basis or depending on the duration of the trial to track subject enrolment· Online reporting of the status of the clinical trial every six months via the SUGHAM portal to know if the trial is ongoing, completed, or has been terminated.
Fee for procuring a license, certificate of registration, and permission for trial
Different fee structures depending on the purpose of the trial. Fees ranging from INR 50,000 to 5,00,000.
Bridging the Gap
The challenges of dealing with clinical trials are multifaceted and involve adhering to the regulatory framework in a responsible and ethical manner by stakeholders, the government, and the judicial system alike.
Patient safety and protection should be of utmost importance, laying strict rules for: 12
Informed consent by audio-visual recording and in a language that the patient is comfortable with
Respect for the patient’s cultural, social, economic, and educational background
Timely reporting of SAEs
New ground rules that can open up the possibility of expanding medical research in India are: 13
Approval of proposals submitted to the DCGI within 30 days of application, if there is no communication from the DCGI
Fast-tracking of domestic approvals
Pre and post-submission meetings with the expert committee to bring in more transparency to the process and to set a well-defined timeline for trial completion
trial compensation in case the investigational drug led to SAEs/death.
A proficient workforce and state-of-the-art infrastructure also play an important role in attracting sponsor companies.
Research has demonstrated that, although Phase III trials are being conducted extensively in India, Phase I trials appear to be limited to the sponsor country.
This could be attributed to the Sponsor’s apprehension in procuring a qualified workforce and technology.
To enable indigenous research in India, it is pivotal to provide personnel with appropriate exposure and continuing medical education, as well as access to up-to-date technology, to be recognized as a country competent enough to conduct all phases of trials. 9
Equally important is the need for skilled healthcare workers to be available nationwide to address the uneven distribution of clinical trials across states.
Concentrating a trial on a particular state could lead to biased conclusions and oversimplify or exaggerate a disease burden or condition.
By providing access to people in all states to join a clinical trial, we not only minimize bias but also include diverse ethnic populations.9
The Future
With positive, patient-friendly, fast-track, and transparent regulatory laws, India will continue to grow as an international hub for testing and developing innovative medicines and medical devices.
Lahiry S, Sinha R, Choudhary S et al. Paradigm Shift in Clinical Trial Regulations in India. Indian Journal of Rheumatology. 2018;13:51-5.
Gogtay NJ, Ravi R, and Thatte UM. Regulatory requirements for clinical trials in India: What academicians need to know. Indian Journal of Anaesthesia. 2017 Mar;61(3):192-9. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5372399/
Burt T, Sharma P, Dhillon S et al. Clinical Research Environment in India: Challenges and Proposed Solutions. Journal of Clinical Research Bioethics. 2014;5:6. DOI: 10.4172/2155-9627.1000201
Chaturvedi M, Gogtay NJ, Thatte UM. Do clinical trials conducted in India match its healthcare needs? An audit of the Clinical Trials Registry of India. Perspectives in Clinical Research. 2017;8(4):172-5.
http://ctri.nic.in/Clinicaltrials/news/CTRI_Newsbulletin_July-Dec_2017.pdf Accessed on April 23, 2019.
Bhave A and Menon S. Regulatory environment for clinical research: Recent past and expected future. Perspectives in Clinical Research. 2017;8:11.6.
Key Highlights of New Drugs & Clinical Trial Rules, 2019. Accessed on April 23, 2019
Dan S, Karmakar S, Ghosh B et al. Digitization of Clinical Trials in India: A New Step by CDSCO towards Ensuring the Data Credibility and Patient Safety. Pharmaceutical Regulatory Affairs: Open Access. 2015;4(3): DOI: 10.4172/2167-7689.1000149.
The information contained in this article is intended solely to provide general guidance on matters of interest for the personal use of the reader, who accepts full responsibility for its use.
Accordingly, the information in this article is provided with the understanding that the author(s) and publisher(s) are not herein engaged in rendering professional advice or services.
As such, it should not be used as a substitute for consultation with a competent adviser. Before making any decision or taking any action, the reader should always consult a professional adviser regarding the relevant article posting.
While every attempt has been made to ensure that the information contained in this article has been obtained from reliable sources, Veeda Lifesciences is not responsible for any errors or omissions or the results obtained from the use of this information.
All information in this article is provided “as is,” with no guarantee of completeness, accuracy, timeliness, or of the results obtained from the use of this information, and without warranty of any kind, express or implied, including, but not limited to warranties of performance, merchantability, and fitness for a particular purpose.
Nothing herein shall, to any extent, substitute for the independent investigations and the sound technical and business judgment of the reader.
In no event will Veeda Lifesciences, or its partners, employees, or agents, be liable to the reader or anyone else for any decision made or action taken in reliance on the information in this article or for any consequential, special, or similar damages, even if advised of the possibility of such damages.
No part of this publication may be reproduced, stored in a retrieval system, or transmitted in any form or by any means, mechanical, electronic, photocopying, recording, or otherwise, without the prior written permission of the publisher.
For information, contact us at:
Veeda Clinical Research Limited
Vedant Complex, Beside YMCA Club, S. G. Highway,
Vejalpur, Ahmedabad – 380 051,
Gujarat India.
Phone: +91-79-3001-3000
Fax: +91-79-3001-3010
Email: info@veedalifesciences.com